Overview
This work studies whether mutation-dependent peptide kinetics can be estimated without exhaustively simulating transition events for every candidate mutation. The approach uses molecular dynamics trajectories sampled within folded and unfolded metastable states to construct a collective variable that captures kinetic sensitivity.
The collective variable is constructed with Harmonic Linear Discriminant Analysis (HLDA) and used within the Collective Variables for Free Energy Surface Tailoring (CV-FEST) framework. The central assumption is that local fluctuations inside metastable states contain information about the free-energy barrier separating those states.
Results
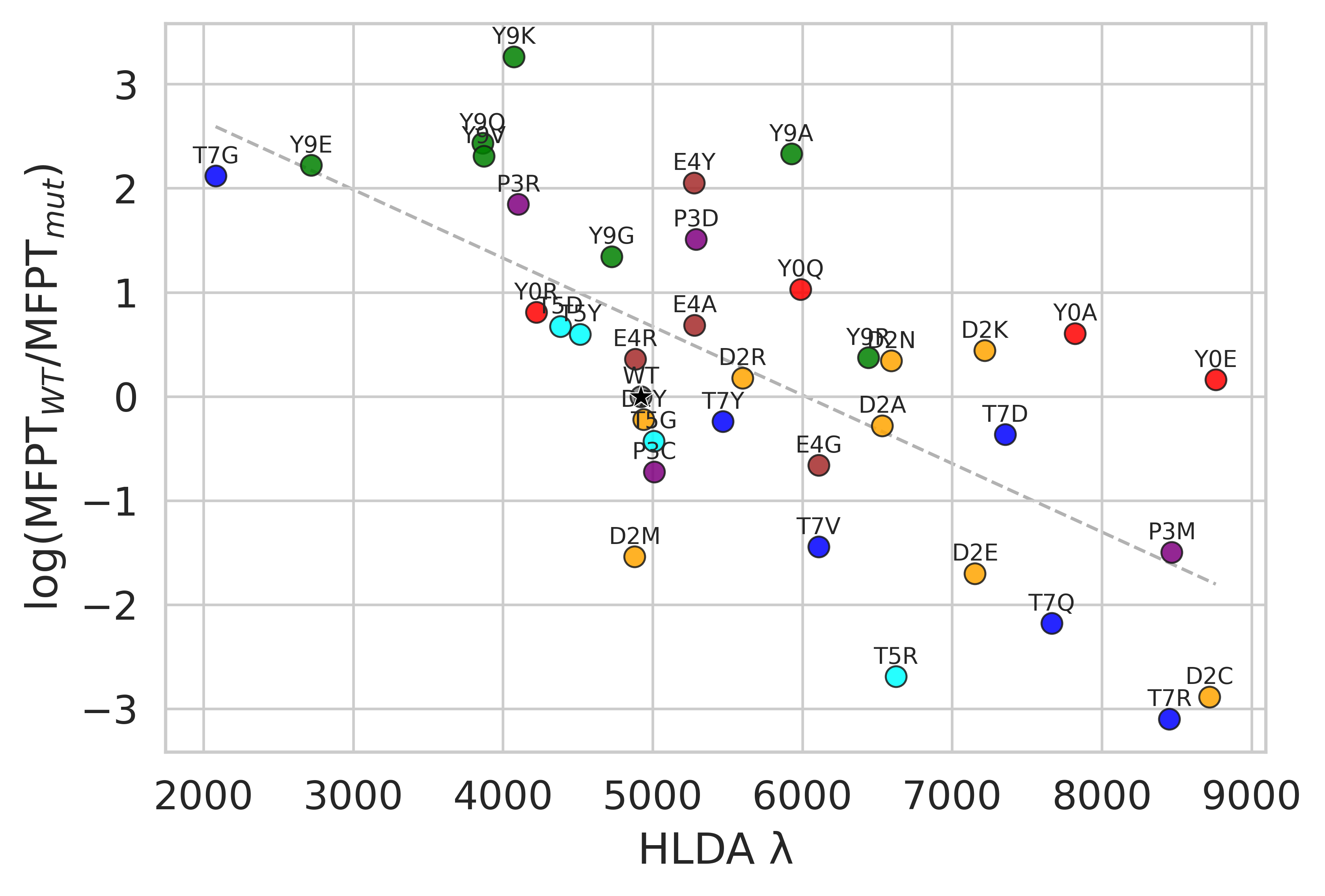
For Chignolin point mutations, the HLDA collective variable derived from the wild-type system produces residue-level scores that identify positions where mutations are expected to accelerate or slow conformational transitions.
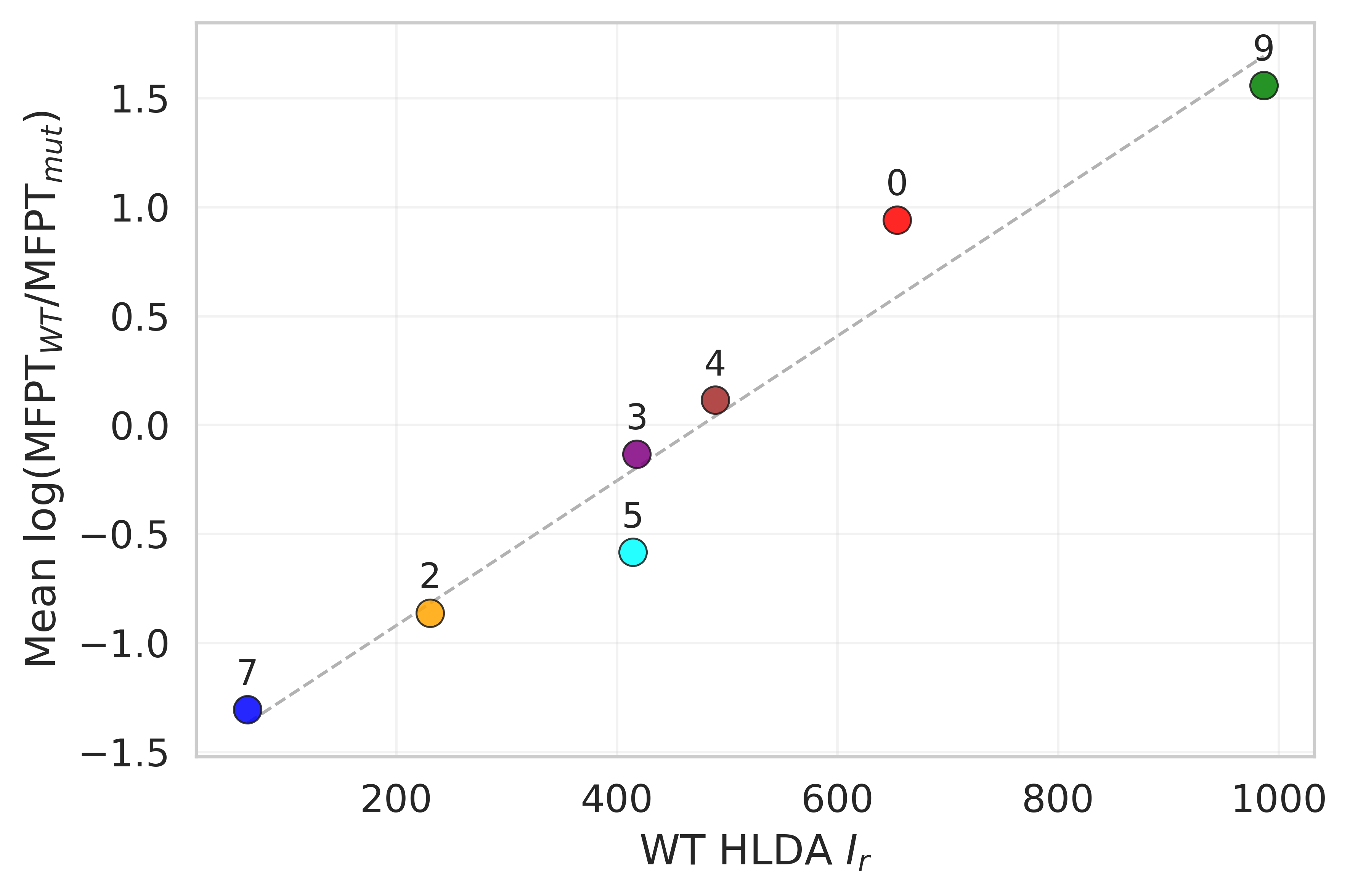
The leading HLDA eigenvalue, which measures one-dimensional statistical separation between folded and unfolded ensembles, also correlates with transition rates across mutations. This makes it a compact descriptor of mutation-dependent kinetic behavior.
Significance
Protein function is shaped not only by structure, but also by thermodynamics and kinetics: the relative stability of states, the height of free-energy barriers, and the rates of conformational transitions.
By estimating mutation effects from limited metastable-state sampling, the method supports a more efficient simulation-driven design loop: propose mutations, estimate their likely kinetic effect, and prioritize candidates before running more expensive rare-event simulations.
Technical Takeaways
The main technical point is not only that HLDA separates folded and unfolded states, but that this separation is predictive. The same low-dimensional description that distinguishes metastable ensembles also carries information about transition-rate changes under mutation.
That connects three things that are often treated separately:
- local equilibrium fluctuations inside metastable states
- free-energy barrier shaping through collective variables
- mutation-dependent conformational kinetics